




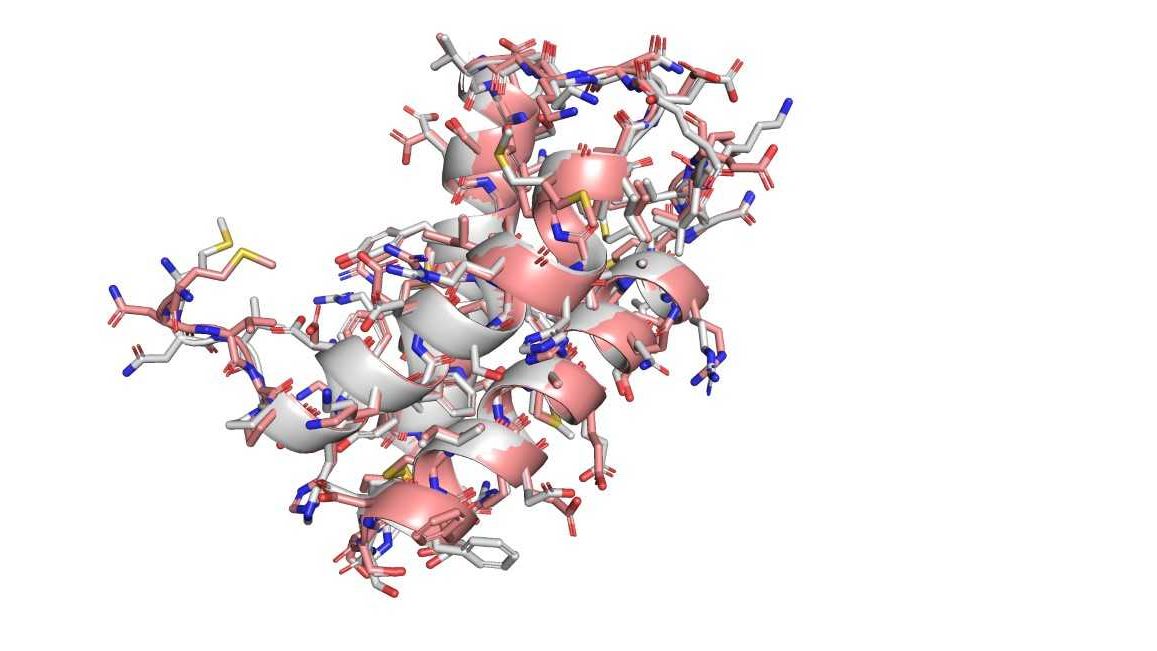
Článek
Prestižní vědecké časopisy, jako je Science nebo Nature, stejně jako vědecké rubriky nejčtenějších zpravodajských serverů, na titulních stranách svých webů informovaly o velkém objevu, který přepisuje dějiny strukturální biologie a zároveň má velký potenciál pro praktické využití. Vědci z celého světa zprávu komentovali slovy jako velký „game changer“, s jehož příchodem v roce 2020 ani nejbližších letech nikdo moc nepočítal.
Co přesně na soutěži CASP (Critical Assessment of Protein Structure Prediction) společnost DeepMind patřící gigantu Google v pondělí oznámila, Seznam Zprávám upřesnil vědecký pracovník z katedry buněčné biologie Přírodovědecké fakulty UK Marian Novotný.

Bílkoviny jsou stavebními bloky života a souvisí s většinou procesů, které probíhají v buňkách. Jak bílkoviny fungují, je přitom determinováno právě jejich tvarem. Zjištění jejich tvaru na základě genetické informace a z ní získaných dat o sekvenci bílkoviny je podle Mariana Novotného velký krok vpřed.
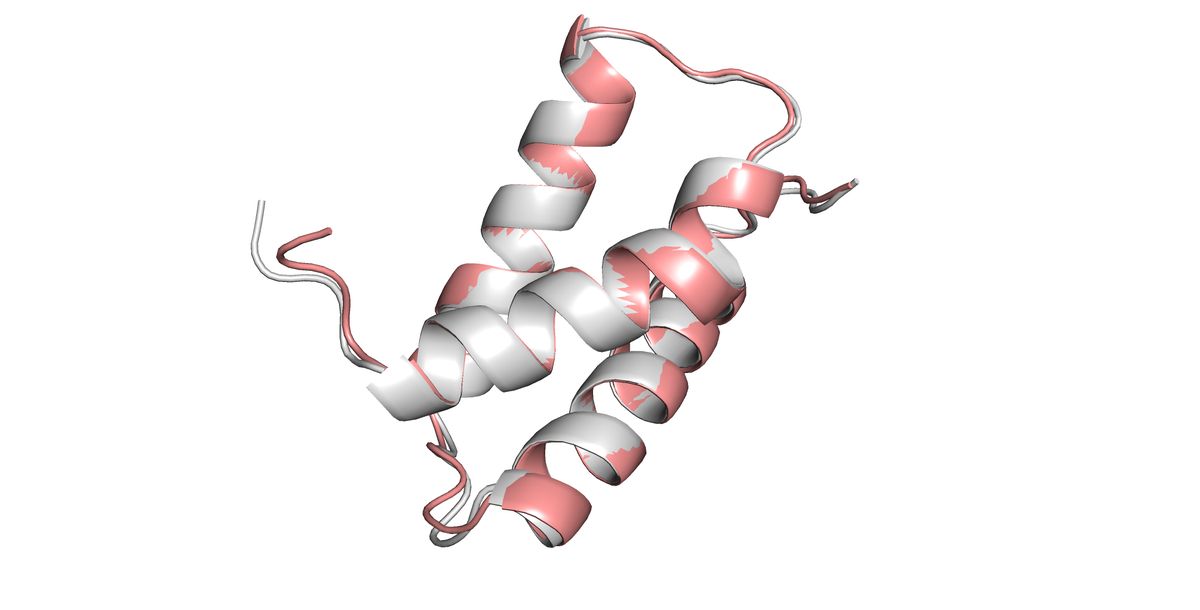
Stejný obrázek jako v úvodním snímku ukazující sekundární struktury bez aminokyselin.
Počítač místo urychlovače částic
„Rozdíl mezi touto metodou a metodami dosud používanými je zásadní. Proteinovou strukturu dnes určujeme třemi hlavními metodami - rentgenovou krystalografií, nukleární magnetickou rezonancí a kryoelektronovou mikroskopií. Všechny tyto metody požadují poměrně složitou instrumentaci,“ uvedl Novotný a dodal, že v nejextrémnějším z jmenovaných příkladů, tedy při rentgenové krystalografii, se musí pracovat s kruhovým urychlovačem částic, synchrotronem, o poloměru půl kilometru, který stojí miliardy a v Evropě jich je jen hrstka. Zbylé metody jsou proveditelné v laboratoři, ale jen s pomocí zařízení v hodnotě stovek milionů korun, drahým provozem a také časovou náročností.
Predikce tvaru bílkoviny postavená na datech ze sekvenování aminokyselin („čtení“ DNA) je naproti tomu mnohem levnější a rychlejší. „Například lidský genom můžete nechat osekvenovat, když to seženete v akci na internetu, za 200 dolarů. Pak můžeme využít toho, že genetická informace je kódovaná pravidly, která my známe, takže pomocí genetického kódu z nukleové kyseliny můžeme prakticky už zadarmo generovat sekvenci aminokyselin v proteinu,“ popsal Novotný, jak jednoduchý je proces dopracování se k datům, která umělá inteligence potřebuje.

„Šít léky na míru“
Otázka, k čemu všemu by nová technologie mohla reálně sloužit, nyní jistě zaměstnává řadu vědců, kteří přemýšlí, jak ji nejlíp uchopit. Už teď je ale podle Novotného jasných několik věcí.
Novotný jakožto vědec zabývající se například 3D modely makromolekul na objevu nejvíc oceňuje potenciál levnější a tím pádem mnohem častější možnosti prohlédnout si tvar proteinů z různých úhlů a v různých velikostech. „Najednou vidíte, že někde je například nějaká kapsa, která by mohla něco vázat, a vy vidíte, jaké aminokyseliny jsou v okolí, a víte, jestli jsou nebo nejsou mutované v některé nemoci,“ přiblížil Novotný důvody, proč se mu díky 3D modelům o bílkovinách lépe přemýšlí.
Technologie ale může podle Novotného skutečně pomoct i ve farmaceutickém odvětví. „Máte třeba protein zodpovědný za nádorové bujení a máte pro něj nějaký inhibitor, načež zjistíte, že přestává fungovat a je potřeba navrhnout jiný, protože člověk je k němu rezistentní, dejme tomu, kvůli další mutaci ve vazebném místě. Když znáte jeho strukturu a víte, jak přesně vypadá, a vidíte, o kterou mutaci jde, tak jste schopní ji namodelovat a jste schopni říct, že se nějaká kapsa v určitém místě třeba o kousek zvětšila. Takže když tam něco přidáte, můžete posílit interakci,“ popsal Novotný na spekulativním příkladu proces, který označil za „šití léků na míru“.

V tuto chvíli ale není jisté, kde bude mít technologie při takovém využití své limity. Problém by podle Novotného mohl nastat u takzvaných bodových mutací, kde dochází k pouze jediné změně v sekvenci aminokyselin. „Ďábel je často právě v detailu a tady to všechno potřebujeme vědět přesně. A my právě ještě nevíme, jestli to takhle přesně umíme. Je možné, že ne,“ řekl Novotný.
Technologie, o které se mluví jako o možném řešení 50 let starého problému proteinového balení, by mohla podle Novotného narazit i u komplexnějších struktur proteinů, kde nemusí obstát. Dalším problémem by mohla být flexibilita proteinů, kterou statický obrázek bílkoviny v jednom konkrétním stavu nezachytí, a nemusí tak úplně stačit pro plné pochopení fungování proteinu.
Jak umělá inteligence funguje, nevědí ani autoři
Umělá inteligence firmy DeepMind nazvaná AlphaFold funguje na principu neuronové sítě, což je výpočetní model inspirovaný lidským nervovým systémem. Novotný fungování popsal jako statistický systém, který pracuje se vstupní informací (sekvencí aminokyselin) a obrovským množstvím dat už známých struktur různých bílkovin, které jsou schraňovány ve veřejných databázích. Ty se plní prostě tím, že jakýkoliv vědec experimentálně zjistí strukturu nějakého proteinu a chce o něm napsat článek, musí do nich data nasdílet. AlphaFold se z těchto dat „učí“, jak struktury vypadají, a díky tomu dokáže poměrně přesně odhadnout jejich tvar.
Co přesně se ale nervová síť naučila, podle Novotného nikdo neví. „Poslouchal jsem přednášku jednoho z autorů tohoto projektu a ani on přesně neví, co se ta neuronová síť naučila. A to je na tom zajímavé. Vy znáte vstup, znáte výstup a jste schopni posoudit, jestli ten výstup je dostatečně dobrý. Ale co vlastně dělá ze vstupu výstup, není zřejmé,“ vysvětlil Novotný.
CASP is both the gold standard for assessing predictive techniques and a unique global community built on shared endeavour. Accuracy is measured on a range of 0-100 “GDT”. #AlphaFold has a median score of 92.4 GDT across all targets - its average error about the width of an atom. pic.twitter.com/cYCN12KxLZ
— DeepMind (@DeepMind) November 30, 2020
Novotný zároveň souhlasí s tím, že příchod takové technologie se nečekal. „Neřeší to všechny problémy světa, ani proteinového balení, ale je to obrovské překvapení. Ten problém se vždycky zdál být strašně komplikovaný. Rovnice, které vedou k jeho vyřešení, jsou tak složité a mají tolik parametrů, že nebyla představa, že by se nám to mohlo povést rozlousknout. Řešení ale kupodivu nejde vůbec přes rovnice, ale přes akumulaci obrovského množství dat, která jsou volně k dispozici.“
Novotný i další vědci oslovení zahraničními médii se shodují, že tohle není konec a dají se očekávat další pokroky, případně odstranění zmiňovaných problémů technologie firmy DeepMind. Ta byla totiž na soutěži CASP jednou ze zhruba stovky dalších projektů, které se predikcí tvaru bílkovin ze sekvence aminokyselin aktivně zabývají. Důležitost soutěže vypíchli i samotní autoři umělé inteligence. CASP funguje už 28 let, odkdy se vědci zabývající se tímto problémem každé dva roky sejdou, poměří síly a vzájemně se obohatí o nové znalosti. Za dva roky se tak možná můžeme těšit na další velké věci.






